Genomic data is expanding at a rapid pace, driven by ambitious efforts to sequence the DNA of millions of species worldwide. Comparative genomics, essentially the science of comparing genomes across species, helps us understand the evolutionary relationships between species. A key part of this is to find homologous regions, which are regions of DNA that are shared across species due to having a common ancestor.
When it comes to homologous genes, there are two main types to know about: orthologs and paralogs. Orthologs are genes that started diverging because of speciation (evolutionary branching into new species), while paralogs diverged because of gene duplication. Orthologs often have similar functions across species, which makes them extremely useful for transferring knowledge from well-studied organisms to newly sequenced ones (Nicheperovich 2022).
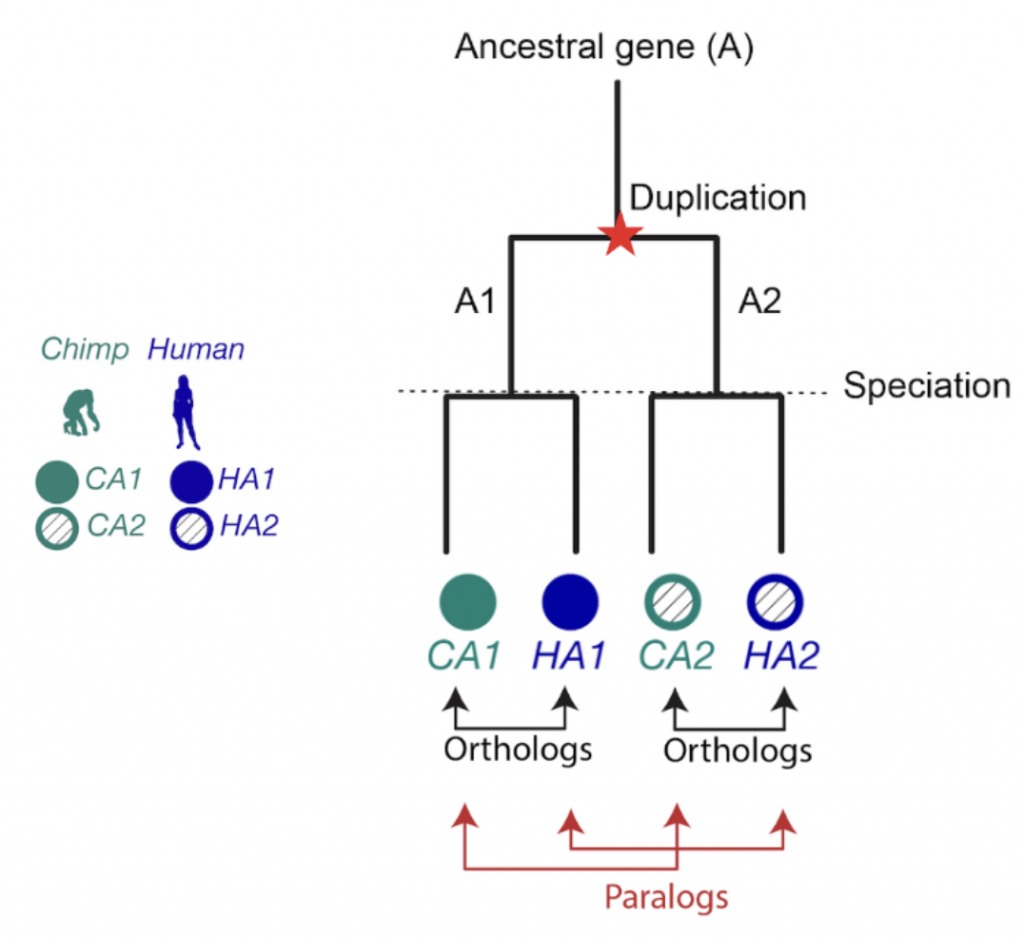
Figure 1. The relationship between two genes that share a common ancestor is called homologous, from the Greek word homologos— homos (meaning “same”) + logos (meaning “relation”). Orthologs are gene pairs that diverged due to evolutionary speciation, while paralogs are gene pairs that diverged due to a duplication event. This distinction is important because orthologs tend to have similar functions, but paralogs do not.
A bit of History!
The idea of distinguishing orthologs from paralogs goes back to Walter Fitch’s seminal work at the University of Wisconsin in 1970 (Fitch 1970). Since then, several research groups have been working on algorithms to accurately estimate orthology. One of the first contributions was the Clusters of Orthologous Groups of proteins (COGs) database, launched by NCBI in 2000, covering 21 genomes of bacteria, archaea, and eukaryotes (Tatusov 2000). More recently, the Orthofinder tool made it possible to find orthologs for a set of genomes of interest with high accuracy. This well-known software uses fast all-against-all gene comparisons with DIAMOND to group genes into orthogroups and refine them with gene trees. Earlier this year, Sonicparanoid presented its second version, which benefits from machine learning to efficiently avoid unnecessary all-against-all alignments, which makes it even faster. All these exciting advancements highlight the thriving community that works in the field of orthology and comparative genomics.
The OMA (Orthologous MAtrix) project came along in 2004 as a method and database for identifying orthologs across genomes (Dessimoz et al. 2005). The original OMA algorithm uses all-against-all gene comparisons with Smith-Waterman to find homologous sequences and then infers orthology relationships from there. Since 2010, Adrian Altenhoff has been the OMA project manager and OMA is hosted at the Comparative Genomics lab, led by Christophe Dessimoz and Natasha Glover. In 2017, Clément Train, a talented PhD student in the lab, took things to the next level with OMA algorithm 2.0, which delivered high precision in orthology inference (Train et al. 2017). Fast forward to today, the OMA Browser has seen 24 major updates where all the orthology data of around 3000 genomes is now presented for easy access with visualization innovations for phylostratigraphy, synteny and gene information (Altenhoff et al. 2024). Along the way, OMA also became a core resource supported by the SIB Swiss Institute of Bioinformatics.
In 2021, I joined the Comparative Genomics lab in Lausanne as a postdoc, took a leap of faith and started working on developing a new algorithm for orthology. The goal was to make it work for several thousands of species, basically scaling to the tree of life—something that’s really needed these days. At first, it felt quite overwhelming as there were several efficient ortholog inference tools such as Panther, OrthoMCL, Orthofinder, Sonicparanoid, Ensembl compara, Domainoid, MetaPhOrs, TOGA and GETHOGS (to name only a few) that are being maintained rigorously and regularly. The developer of these tools made great contributions to the field, and the huge number of comparative genomics studies over the years wouldn’t have been possible without these softwares. Their intricate design and comprehensive algorithms are accurate and efficient, making it hard to imagine advancing the field even further.
On top of that, I was new to the field—my PhD was on diploid and polyploid haplotype phasing using DNA sequencing reads (Majidian et al. 2020) and my background is in engineering and signal processing. But, I embarked on this journey and started learning concepts and methods in comparative genomics. I was lucky to have great mentors and lab mates who were always open to answering my questions, over zoom and in-person.
OMA turns young!
Let’s talk about FastOMA. With contributions from several lab members (Stefano, Yannis, Ali, Alex, David) and guidance from Christophe, Adrian and Natasha, we developed and implemented the FastOMA method. FastOMA works by benefiting from the current knowledge of orthology available on the OMA browser. FastOMA first maps the input genes (at amino-acid level) to reference gene families (the Hierarchical Orthologous Groups, HOGs), using OMAmer, a fast k-mer-based mapper. To learn about HOG, see this YouTube video by Natasha. Next, FastOMA works on each family separately. In other words, FastOMA does not perform comparison of genes from one family to another since these genes do not have any shared homology. This is an important step which saves us a huge amount of computations. Then, FastOMA infers the gene trees on (a subsample of) genes at each taxonomic level to distinguish orthologs from paralogs within each family. This phylogeny-guided subsampling is also key to maintaining speed and accuracy at the same time.
FastOMA’s speed makes it possible to handle genomic datasets with thousands of species. FastOMA uses the “OMA’s knowledge”, and is now swift as OMA turns young. FastOMA achieves high accuracy and resolution, as shown by the Quest for Orthologs benchmarks (Majidian, 2024).
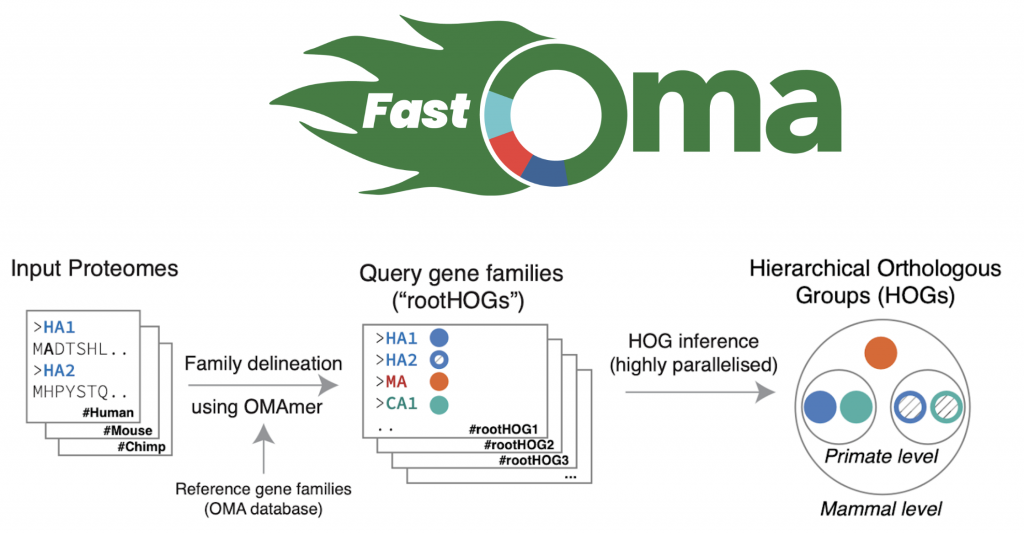
Figure 2. Overview of how FastOMA infers orthologs.
To the future!
As a community, we work collaboratively to advance the field and the lab has been contributing to the benchmarking datasets, making it possible to compare the performance of different tools, and ultimately advance the field. Earlier this year, in July, the Quest for Orthologs event (QFO8) was held at the University of Montreal, where recent advancements in orthology inference were discussed, and FastOMA was also presented there. The QFO 9 will be in Switzerland in 2026!
There are several directions for improving FastOMA’s accuracy and speed further. One exciting direction is taking advantage of recent advancements in protein structure prediction to reconstruct structural trees (Moi et al. 2023) in the context of orthology inference. This could really help boost resolution at deeper evolutionary levels. Besides, it would be very interesting to use gene order conservation, a.k.a, synteny information (Bernard et al. 2024), which could serve as an additional layer of information to refine orthology predictions. We hope our proposed hierarchical approach accompanied with several ideas will stimulate further developments.
So far, FastOMA has caught the attention of several labs around the world, who incorporated FastOMA in their studies. We are excited to hear how you plan to use FastOMA into your own research. Feel free to create a GitHub issue (https://github.com/DessimozLab/FastOMA) or send us an email if any help is needed!
To learn more see FastOMA academy: https://omabrowser.org/oma/academy/module/fastOMA
This post was originally published in Nature Communities here.
References
- Altenhoff, Adrian M., et al. “OMA orthology in 2024: improved prokaryote coverage, ancestral and extant GO enrichment, a revamped synteny viewer and more in the OMA Ecosystem.” Nucleic Acids Research 52.D1 (2024): D513-D521. doi:10.1093/nar/gkad1020
- Bernard, Charles, et al. “EdgeHOG: fine-grained ancestral gene order inference at tree-of-life scale.” bioRxiv (2024): 2024-08. https://doi.org/10.1101/2024.08.28.610045
- Dessimoz, Christophe, et al. “OMA, a Comprehensive, Automated Project for the Identification of Orthologs from Complete Genome Data: Introduction and First Achievements” RECOMB 2005 Workshop on Comparative Genomics, LNCS 3678 (pp. 61-72). link
- Emms, David M., and Steven Kelly. “OrthoFinder: phylogenetic orthology inference for comparative genomics.” Genome Biology 20 (2019): 1-14. doi:10.1186/s13059-019-1832-y
- Fitch, Walter M. “Distinguishing homologous from analogous proteins.” Systematic zoology 19.2 (1970): 99-113. doi:10.2307/2412448
- Majidian, Sina, et al. “Orthology inference at scale with FastOMA.” Nature Methods (2025) doi:10.1038/s41592-024-02552-8
- Majidian, Sina, Mohammad Hossein Kahaei, and Dick de Ridder. “Minimum error correction-based haplotype assembly: Considerations for long read data.” PLOS ONE 15.6 (2020): e0234470. doi.org/10.1371/journal.pone.0234470
- Moi, David, et al. “Structural phylogenetics unravels the evolutionary diversification of communication systems in gram-positive bacteria and their viruses.” BioRxiv (2023): 2023-09. doi:10.1101/2023.09.19.558401
- Nicheperovich, Alina, et al. “OMAMO: orthology-based alternative model organism selection.” Bioinformatics 38.10 (2022): 2965-2966. doi:10.1093/bioinformatics/btac163
- Tatusov, Roman L., et al. “The COG database: a tool for genome-scale analysis of protein functions and evolution.” Nucleic acids research 28.1 (2000): 33-36. doi:10.1093/nar/28.1.33
- Train, Clément-Marie, et al. “Orthologous Matrix (OMA) algorithm 2.0: more robust to asymmetric evolutionary rates and more scalable hierarchical orthologous group inference.” Bioinformatics 33.14 (2017): i75-i82. doi:10.1093/bioinformatics/btx229




