
The problem in a nutshell
Quantitative Trait Loci (QTL) are regions of a genome for which genetic variants correlate with particular traits. To take a simple example in plants, one might observe that the average seed size (trait) is significantly larger when considering the subset of a population which has a C at a particular position in the genome than a subpopulation with a T.
The reason QTL identifies genomic regions and not precise positions is that neighbouring variants tend to be inherited together. These regions typically contain hundreds of genes, making it difficult to say which one(s) are causal to the trait variation—if any at all (the causal genetic variation(s) can be in non-coding regions too).
Thus, to prioritise candidate causal genes within a QTL region, researchers typically consider previous knowledge on these genes, to see whether a particular gene “makes sense”. In the case of seed size, it might be a gene previously implicated in growth or regulation, or a gene known to influence seed size in a different species. This process is however requires substantial manual interpretation, and is thus labour-intensive and haphazard.
Enter QTLsearch
We realised that our framework of hierarchical orthologous groups, which relates genes across many species, could be extended to integrate QTL results with previous gene function annotations.
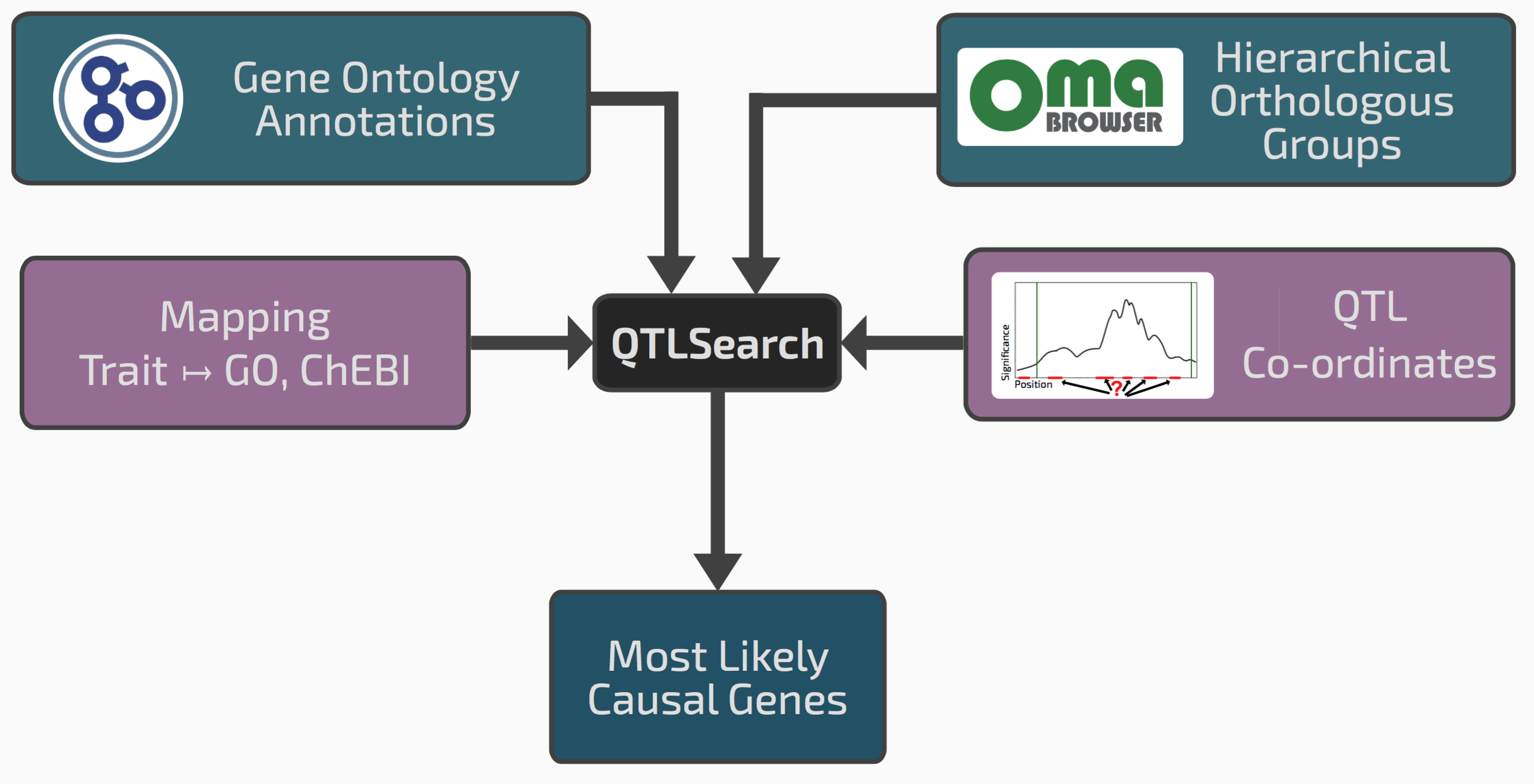
Conceptual overview of QTLsearch
If we go back to the seed size example, it might be that among the genes in the window, one has an ortholog in a different species previously annotated with the GO term “reproductive system development”. This could be a good candidate causal gene.
One risk however in integrating lots of previous knowledge across many species is that we might also find some spurious patterns. We therefore had to devise a way of controlling for random associations between QTL regions and evolutionarily propagated knowledge. Such “null distribution” depends on the specificity or the terms in question, the amount of annotations, the size of the QTL regions, and the species sampling. To cope with this complexity, we chose to implement a non-parametric permutation test.
We implemented the tool as an open source package called QTLsearch, available here.
QTLsearch infers more candidate causal genes than manual analyses
We used QTLsearch to reanalyse two previous studies. In both cases, we could call more candidate genes than the original studies. But more importantly, the evidence behind our calls is fully traceable and statistically supported.
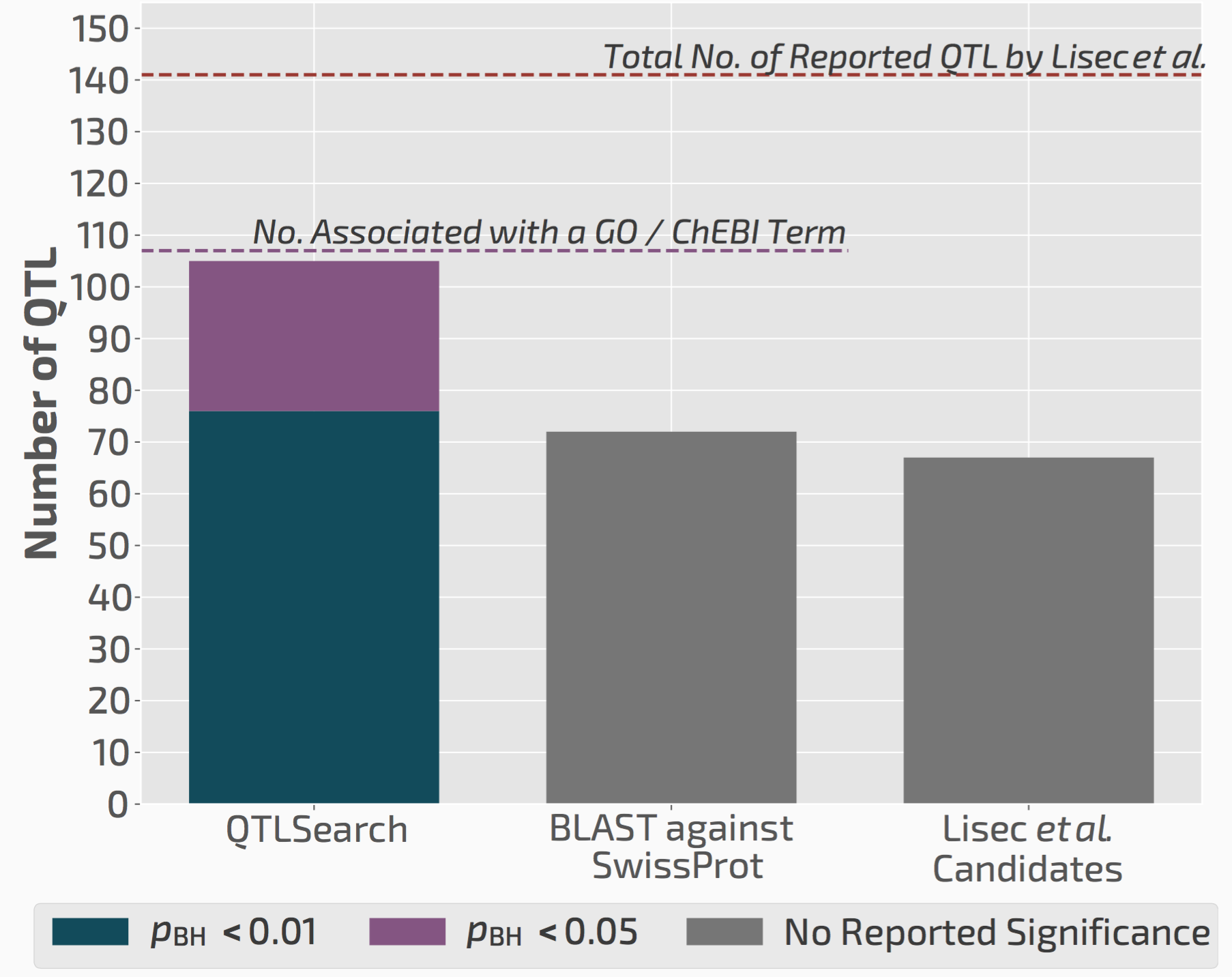
QTLsearch could identify more candidate genes than the original study, but in an automated, reproducible, and statistically meaningful way.
Thus we think this will greatly facilitate future QTL analyses, particularly those that are done in non-model species for which the previous experimental knowledge is very limited.
Behind the paper
This is the third paper that resulted from our collaboration with Bayer CropScience (now BASF CropScience), after our work on homoeologs and on detecting split genes.
The project was conceived by Henning Redestig, collaborator at Bayer at the start of the project (now at DuPont). Henning had contributed to a QTL study and knew how labor intensive the search for putative causal genes is. He realised that HOGs could provide a natural way of integrating functional knowledge across multiple species, to combine the QTL information with previous functional data.
Alex Warwick Vesztrocy, PhD student on the project and first author, ran with the idea—promptly implementing and testing it. Early results looked promising, but Alex soon realised that the mapping between metabolites and GO terms could be improved. He also realised that some terms were quite common, so he devised the approach to compute the significance scores.
Our manuscript was accepted as proceedings paper at the European Conference on Computational Biology (ECCB). In our lab, we like proceedings paper. It’s nice to be able to present the work and publish the paper, particularly since the ECCB proceedings appear in a good journal. More importantly, conferences impose hard deadlines. Deadlines for submission of course, but also for peer-reviewing and for deciding acceptance or not!




